Introduction to bash language for bioinformatics
Introduction
The difficulty of learning bash is often underestimated by others, who expect people approaching bioinformatics to learn it automatically. Here we try to put together the first basic concepts and commands.
Using the bash command line becomes quickly essential if you are doing bioinformatics.
First of all, you might need it to access a computing cluster (for example, GenomeDK at Aarhus University), since most clusters runs on a UNIX-based operating system, such as Linux, using a bash command line.
Just as important is the fact that on a command line you can very easily do operations on multiple and very large files, something you would not be able to do using, for example, R or python. Large sequences of operations can be automatized into pipelines (an advanced topic not for this tutorial).
With a command line you can run many small programs, compose them together, and organize them in a chain of commands. This type of program organization fits well with what a bioinformatics project consist of: many tools to be applied repetitevely on multiple large files, and organizing those programs in a specific sequence. An example could be aligning to a reference genome many raw bulk-RNA sequencing files: the alignment operation must be repeated many times, and when files are finished, they might need to be merged if they are from the same sample.
Some terminology
When using a UNIX operating system (Linux, MacOs), everything on your computer fits one of two categories: processes and files.
- Processes are running instances of a program, and a program is any executable file stored in your computer.
- A file is any collection of data (program, image, video, audio, …).
The terminal is a text-based interface where you can write commands in various languages, depending on your choice. A terminal looks rather primitive, but it is what you often use on a daily basis to perform bioinformatics operations or control computing clusters.
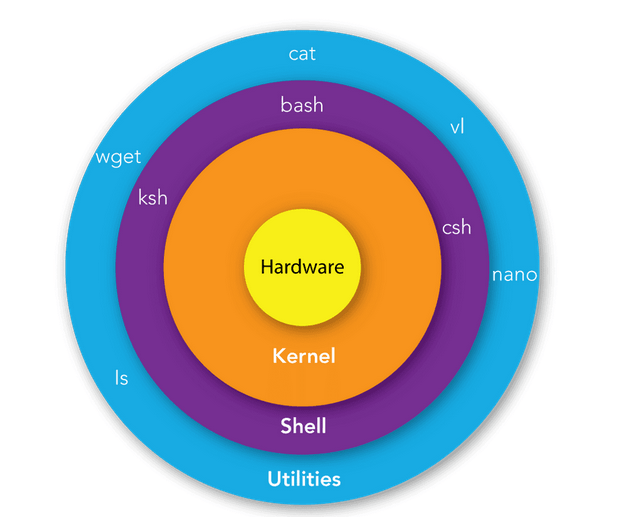
Whenever we write a command on the terminal and press enter, we have a shell receiving the code we wrote through the terminal. The shell is the outer layer of the operating system, which inteprets the commands and communicates them to the kernel.
The kernel is the core of the operating system, managing the computer physical components (hardware) and interfacing them with the processes that need to run. In general, any program (browser, game, …) you open or action (moving files, renaming folders, …) you do on your computer ends up being a process managed by the kernel. This communication process is shown in Figure 1
Efficiency and Speed
We can roughly identify various levels of efficiency, manual work, speed, number and size of handled files when working with a command line, the typical languages like R and python, or bash pipelines:
| Programming mode | Nr of files | File Size | operational speed | Manual work |
|---|---|---|---|---|
| R, python, … | from 1 to 10s | small | package-dependent | A lot |
| Command line | from 1 to 100s | 1-10s GB | fast | Low-moderate |
| Unix Pipeline | from 1 to many 1000s | many TB | fast | Low |
You will see in this tutorial how we can use basic bash utilities and handle text files. Those files would take longer time to read in R and python and the code to modify them would be in general longer and slower.
Both the bash language and python or R are interpreted languages. You shouldn’t consider them as opposed to each other, but rather as complementary. Bash can do many things python or R cannot do very efficiently: search for files, look for patterns in long text files, concatenate operations, create pipelines, be the preferred system language on computing clusters. Python or R are great to perform for example statistical analysis.
So the best way to approach all those languages is to harness their advantages, instead of consider one of them “worse” than the others.
What is Bash? Help you with this link if necessary
Slides
Our slides introducing the bash shell
Tutorial
Here starts the tutorial. There is only one technical prerequisite, that is, you need a Terminal. The terminal is where you can write your commands - which are then sent to the computer to be executed.
Mac and Linux computers already have a software called
Terminalinstalled (they are both computers with UNIX-based operating systems)Windows have a different sort of terminal called Powershell (it is DOS-based and not UNIX-based). Please install
MobaXTerm, then open it and click Start Local Terminal.
Terminal and folders anatomy
When you work in the terminal, you will always see a prompt which starts with something of this type

which provides you
- username (e.g. samuele)
- computer name (e.g. D55749)
- current working directory where the user is working at the moment (e.g.
~, which is the short form for the home directory)
In MobaXTerm you see only date, time and current working directory highlighted with various colors.
Home directory ~
The home directory, which can be written as ~, is usually of the form /home/username, and is private to the user (no other users of that computer can access it).
When you open the terminal, you always start with current working directory as your home. Try to write and execute (pressing enter) the command
pwdand you will see the full home path (which is the folder structure leading to your home directory)
Current working directory (cwd)
Every command you execute refers to your cwd. For example, write
lsand you will see the list of files in your cwd. Try to create an empty file now with
touch emptyFile.txtand create a folder, which will be inside our cwd:
mkdir myFolderIf you use again the ls command, the new file and folder will show in the cwd.
Now we want to download something from the internet, for which we have a download link. We are getting a raw sequenced dataset in fastq format, which is currently gzip-compressed. The wget command can be used for the download. Note that now we also add an option -O to provide the output file name as ./myFolder/data.fastq.gz, where the dot . stands for the cwd, followed by myFolder, followed by the file name.
wget https://github.com/hartwigmedical/testdata/raw/master/100k_reads_hiseq/TESTX/TESTX_H7YRLADXX_S1_L001_R1_001.fastq.gz -O ./myFolder/data.fastq.gzNot all utilities are installed in MobaXTerm. If you get an error, install wget with the command
apt-get -y install wgetand then try the download again.
Paths and navigating the directory tree
We have already been using directories and paths a lot, so it is time to polish some definitions. Files are organized in a directory tree, which restricted to the tutorial we are running looks like Figure 2. Here home is a root folder, which is one of the top-level folder of your computer, so it has / at the beginning of its name.
/home contains a folder with your username, which is right now your cwd. Inside your cwd you have the empty file and a directory myFolder containing the data.
Directories and files are organized with a tree hierarchy, so that /home is the first level, username the second, and myFolder the third level. The path in the directory tree to data.fastq.gz is expressed as /home/username/myFolder/data.fastq.gz.
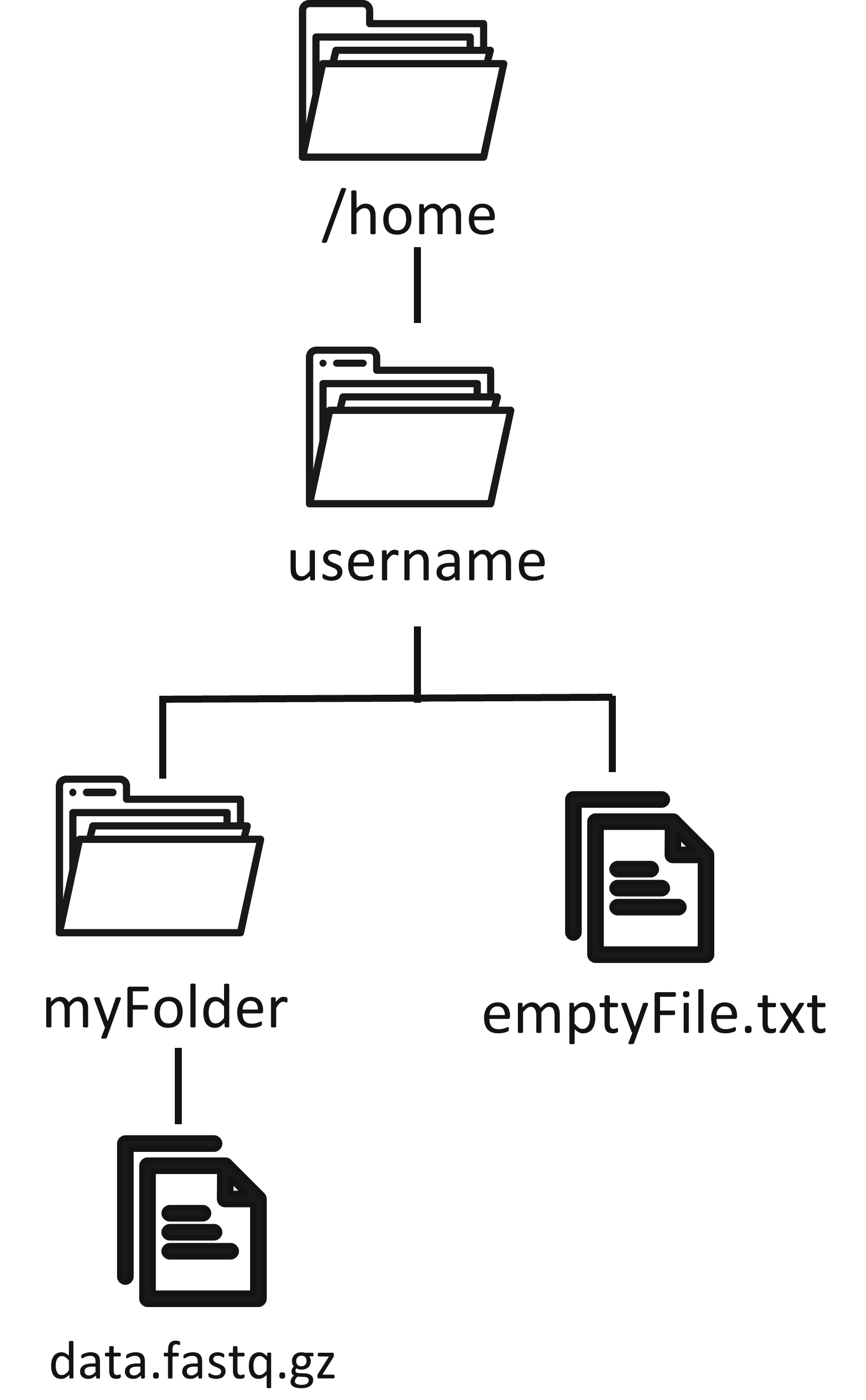
/ symbol. Other folders and files are at subsequent branch levels of the root folder /home. Some folders specific to your computer might be missing from this scheme.
Absolute and relative path
The path /home/username/myFolder/data.fastq.gz is called absolute because independent of your cwd. Let’s try another absolute path: the root folder /usr contains all executable files of the bash utilities we are using in this tutorial. Such files are in the folder bin. Execute
ls /usr/bin/The output is a long list of executable files and some folders (sometimes they also have different colors, depending on your terminal settings). If you scroll and look, you can find familiar names like wget, ls, and so on. Now, the path /usr/bin is independent of our cwd.
On the contrary, the path ./myFolder/data.tar.gz depends on the cwd, and is equivalent to write myFolder/data.tar.gz, because ./ is always included by default (. represents the cwd). All paths that do not start with a root folder are relative!. To look inside myFolder, we can write
ls myFolder/How do you write the command above using absolute paths? Remember that ~ correspondes to /home/username
Working with (text) files
Many files you use in bioinformatics are nothing else than text files which are written in a specific matter. This specific way of arranging the text in the files gives you many of the file formats you encounter when doing bioinformatics.
Some file formats are encoded differently than with plain text, and cannot usually be seen with a text editor.
Less for reading files
The first thing you want to do, is to check at how the content of a file looks like. The program less is perfect for this purpose: you can scroll text with the arrows, and use the keyboard to do operations like searching for text or jumping to a line. Try
less data.fastqand see the format of your data, where each four lines provide a sequence. The very first sequence you see should be
@HISEQ_HU01:89:H7YRLADXX:1:1101:1116:2123 1:N:0:ATCACG
TCTGTGTAAATTACCCAGCCTCACGTATTCCTTTAGAGCAATGCAAAACAGACTAGACAAAAGGCTTTTAAAAGTCTA
ATCTGAGATTCCTGACCAAATGT
+
CCCFFFFFHHHHHJJJJJJJJJJJJHIJJJJJJJJJIJJJJJJJJJJJJJJJJJJJHIJGHJIJJIJJJJJHHHHHHH
FFFFFFFEDDEEEEDDDDDDDDDHere the first line is informative of the sequence, the second is the sequence itself and then you have an empty line (symbol +), followed by the quality scores (encoded by letters according to this table).
Try to scroll in all directions (with the arrow keys) to explore some lines in the file. If you want to exit from the text viewer, press q.
Try to search online how to look for a specific word in a file with less. Then visualize the data with less, and try to find if there is any sequence ten adjacent Ns (which is, ten missing nucleotides). Then, answer the question below
How long is the shortest sequence of missing nucleotides not found anywhere in the file data.fastq?
Usually, a bash shell command can be found by searching for the command + man, for example less man. The manual of less lists all the many (many many) functionalities that can be useful to explore text files, which is always a good activity to do for double checking if things are in order.
In general: always check the manuals or write on the prompt the option --help (such as less --help). Usually you will understand a lot of what you can do and how from that.
File properties
How big is a file? How many lines does it have? When has it been created? You can use the good old ls with some extra options to list all files with some extra info:
ls -lah You should get something like this depending on your computer:
totalt 6,3M
drwxr-xr-x+ 1 au612681 UsersGrp 0 28 aug 11:38 .
drwx------+ 1 au612681 UsersGrp 0 28 aug 10:38 ..
-rw-r--r--+ 1 au612681 UsersGrp 6,3M 28 aug 11:38 data.fastqthe last line is clearly the data file, with creation date and size. Other infos about the file are group and user ownership and which rights we have on the file (we will not talk about these in this tutorial).
Look also at the other two elements: . and .., which represents the current directory and the one above in the directory structure. Those link you to the directories, so one can always change to the directory above by using cd ..
You can combine multiple times .. to change directories multiple levels above. Use the terminal and execute the command cd ../../. Which folder does it bring you to?
Oh damn, now we changed cwd, and we cannot remember the path to the previous one where we have the data. We can use -, which brings us to the previous cwd!
cd -Now, how many lines are there in your file? The command wc can show that to you. It has many options, but we are going to use the one to count lines in a file. As always, look for the manual or examples to see how you can use it in other many ways.
wc -l data.fastqwill tell you that the file has 100000 lines, so you have 25000 sequences (each sequence is defined by 4 lines).
Copy and Move
Now we learn a few useful commands because we want to work on more than one file. We will create multiple copies of our file. To make a copy of the data file and called the new one dataCopy.fastq, do
cp data.fastq dataCopy.fastqThen we will move data.fastq. You could move it in any folder, even in the current directory. If you move it into the current directory, you can use the mv command to simply change its name:
mv data.fastq dataOriginal.fastqIf you now use ls -lah, you will see that you have two files of identical size and different creation dates. But… are those files identical? diff can tell you if you provide two file names. It will print out differences (or nothing if files are identical)
diff dataOriginal.fastq dataCopy.fastqWriting on a file
You can write something on a file using >. This writes any output from a command which would appear on the screen into a file. For example the following commands prints out the first four lines of the data, but they will end up inside the file fourLines.fastq
head -4 dataOriginal.fastq > fourLines.fastqTry to use
cat fourLines.fastqto see those four lines printed out directly on the screen from the file.
Using again > to write on the same file will not add text to the file, but will overwrite it with the new text.
Let’s append the first four lines of the data to its copy without overwriting it. This can be done using >>:
head -4 dataOriginal.fastq >> dataCopy.fastqWe want to run again the diff command to compare the two sequence files. Do you need to rewrite it? Of course not! Your bash saves the command history.
Keep CTRL (or CMD on Mac) pressed, and in the meanwhile press r, then start writing diff. The old command will show up and you can run it pressing Enter. Alternatively, press the arrow UP to see the history of commands backward.
If you run the diff command on the two files you will see an output. This shows quite a lot of information and not only the sequence which is different:
--- dataOriginal.fastq
+++ dataCopy.fastq
@@ -99998,3 +99998,7 @@
means that there are additional lines in the second file (+++), missing in dataOriginal.fastq (- - -), and their coordinates.
Piping
A last important concept is the pipe. You can create small pipelines directly on the shell with the symbol |. Every time you use the pipe symbol, you get the output of a command and send it to the next command. For example, the grep command can find a pattern in a file in this way
grep NNNNN dataOriginal.fastqNow, what if we want to find that pattern in the first hundred sequences? Easy! We use head, and then pipe it into grep! When doing this, you do not need to provide a file to grep, and in that way it will read the output of the pipe.
head -400 dataOriginal.fastq | grep NNNNNThat was cool! In this case we have a small output on screen, but the output can be really huge as well. Maybe it makes sense to count how many sequences grep can find. Well, can’t really do it by hand! Pipe the result once more using wc as we did earlier!
head -400 dataOriginal.fastq | grep NNNNN | wc -lWait a second! That sequence absolutely needs to be saved into a file. How do you do it!
Shortcuts and useful keys combinations
This was all for this very introductory tutorial. You have already seen how the bash shell is flexible, though requiring a bit of a steep learning curve.
We want to recap here useful shortcuts and key combinations for your day-to-day use of the bash shell
Here’s a list of useful shortcuts and key combinations that you can use in the Bash shell to improve your efficiency:
Editing Text
Ctrl + U: Delete the text from the cursor to the beginning of the line.Ctrl + K: Delete the text from the cursor to the end of the line.Ctrl + W: Delete the word before the cursor.Alt + D: Delete the word after the cursor.
Command Completion and Expansion
Tab: Auto-complete file names, directory names, or commands.Alt + .: Use the last argument of the previous command in the current command line.
Other Useful Shortcuts
Ctrl + C: Terminate the current command (useful if things are stuck).Ctrl + D: Log out of the current shell (equivalent toexit).Ctrl + T: Swap the last two characters before the cursor.
Now you are able to navigate the directory tree and visualize text files with less. You can also perform some basic operations, including piping commands, which is useful when handling large outputs which might end up directly on your screen.